Robert J Allaway 0000-0003-3573-3565
· allaway
· allawayr
Sage Bionetworks
· Funded by Neurofibromatosis Therapeutic Acceleration Program; Children’s Tumor Foundation
Deepashree Venkatesh Prasad 0000-0001-5756-4083
· dvenprasad
Childhood Cancer Data Lab, Alex’s Lemonade Stand Foundation
Justin Guinney 0000-0003-1477-1888
· jguinney
Sage Bionetworks
· Funded by Neurofibromatosis Therapeutic Acceleration Program; Children’s Tumor Foundation
Casey Greene✉ 0000-0001-8713-9213
· cgreene
Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania; Childhood Cancer Data Lab, Alex’s Lemonade Stand Foundation
☯: These authors contributed equally to this work.
(Instructions: Describe the background, basic structure of the article, list material to be covered indicating depth of coverage, how they are logically arranged, include recent pubs in the area, 300-500 words)
The advent of high-throughput profiling methods such as genomics, transcriptomics, and other technologies has accelerated basic research and made deep characterization of patient samples routine.
These approaches provide a rich portrait of genes, cellular pathways, and cell types involved in complex phenotypes.
Machine learning is often a perfect fit for extracting disease-relevant patterns from these high dimensional datasets.
Often, machine learning methods require many samples to identify recurrent and biologically meaningful patterns.
With rare diseases, biological specimens, and consequently data, are limited due to the rarity of the condition.
In this perspective, we outline the challenges and emerging solutions for using machine learning in rare disease settings.
We also note that precision medicine presents a similar challenge, in which a common disease is partitioned into small subsets of patients with shared etiologies and treatment strategies.
Advances from rare disease research are likely to be highly informative for other applications as well, and we propose that the methods community should prioritize the development of machine learning techniques for rare disease research.
Introduction
Rare disease research is increasingly dependent on high-throughput profiling of samples and would greatly benefit from machine learning (ML) analytics.
Machine learning algorithms are computational methods that can identify patterns in data, and can use information about these patterns to perform tasks (e.g., pick out important data points or predict outcomes when they are not yet known, among other tasks).
A systematic review of application of ML in rare disease uncovered 211 human data studies that used ML to study 74 different rare diseases over the last 10 years [1].
Indeed, ML can be a powerful tool in biomedical research but it does not come without pitfalls [TODO: ref], some of which are magnified in a rare disease context.
In this perspective, we will focus our discussion on considerations for two types of ML in the context of the study of rare diseases: supervised and unsupervised learning.
Supervised algorithms require training data with specific phenotype labels (e.g., responder vs. non-responder) and learn correlations of features with the phenotype labels to predict the phenotype labels of unseen or new test data (e.g., predicting which new patient would or would not respond to treatment).
If the goal of a study is to classify patients with a rare disease into molecular subtypes based on high-throughput profiling, researchers would select a supervised ML algorithm to carry out this task.
A supervised ML model is of limited utility if it can only accurately predict phenotype labels in the data it was trained on (this is called overfitting); instead, it’s more beneficial to develop models that generalize or maintain performance when applied to new data that has not yet been “seen” by the model.
In later sections, we’ll discuss regularized models, a strategy for reducing overfitting that can be useful for rare disease datasets.
Unsupervised algorithms can learn patterns or features from unlabeled training data.
Examples of unsupervised learning include principal component analyses (PCA), k-means or hierarchical clustering, or t-distributed stochastic neighbor embedding (t-SNE).
In the absence of known molecular subtypes, unsupervised ML approaches can be applied to identify groups of samples that are similar and may have distinct patterns of pathway activation [TODO: ref].
Unsupervised approaches can also extract combinations of features (e.g., genes) that are indicative of a certain cell type or pathway.
Often, too few samples (not enough data) leads to challenges in successfully training a model, or in identifying signals that are useful for biological discovery.
Though researchers strive to train useful and informative models, there are challenges inherent to applying ML to rare disease datasets.
For example, training supervised models requires datasets where the phenotype labels have very little uncertainty (or “label-noise”) [2] – termed “gold standard” datasets – but rare disease datasets often come with significant label-noise (e.g., silver standard datasets) due to limits in the current understanding of underlying biology and evolving clinical definitions of many rare diseases.
Label-noise can decrease prediction accuracy and require larger samples sizes during training [3].
ML methods also benefit from using large datasets, but analyzing high dimensional data from rare diseases datasets that typically contain 20 to 99 samples is challenging [1,4].
Small datasets lead to a lack of statistical power and magnify the susceptibility of ML methods to misinterpretation and unstable performance.
While we expect ML in rare disease research to continue to increase in popularity, specialized computational methods that can learn patterns from small datasets and can generalize to newly acquired data are required for rare disease applications [5].
In this perspective, we first highlight ML approaches that address or better tolerate the limitations of rare disease data, and then discuss the future of ML applications in rare disease.
High-throughput ‘omic’ data methods generate high-dimensional data or data with many features, regardless of the underlying disease or condition being assayed.
A typical rare disease dataset is comprised of a small number of samples [1].
A lack of samples gives rise to the “curse of dimensionality” (i.e., few samples but many features), which can contribute to the poor performance of models [6] [TODO: reference new figure as appropriate #186].
More features often means increased missing observations (sparsity), more dissimilarity between samples (variance), and increased redundancy between individual features or combinations of features (collinearity) [7], all of which contribute to a challenging prediction problem.
If a small sample size compromises an ML model’s performance, then two approaches can be taken to improve the outcome: 1) increase the number of samples to reduce sparsity, variance, and collinearity, 2) improve the quality of samples to account for sparsity, variance, and collinearity
In the first approach, appropriate training, testing, and validation sets could be constructed by combining multiple small individual rare disease cohorts [TODO: Link to experimental design box #185].
In fact, this is often required for the study of rare diseases in the authors’ experience.
In doing so, special attention should be directed towards harmonization since data collection can differ from cohort to cohort.
Without careful selection of aggregation methods, one may introduce technical variability into the aggregated dataset which can negatively impact the ML model’s ability to learn or detect meaningful signal.
Steps such as reprocessing the data using a single pipeline, using batch correction methods [8,9], and normalizing raw values [10] may be necessary to mitigate unwanted technical variability.
In the second approach, small but meaningfully generated datasets can greatly enhance the performance of ML models in the context of rare disease.
Specifically, improving labeling of data is critical in accounting for sparsity and variance in the data.
In our experience, collaboration with domain experts has proved to be critical in gaining insight into potential sources of variation in the datasets.
An anecdotal example from the authors’ personal experience: conversations with a rare disease clinician revealed that samples in a particular tumor dataset were collected using vastly different surgical techniques (laser ablation and excision vs standard excision).
This information was not readily available to non-experts, but was obvious to the clinician.
Addition of this kind of important metadata or labels to the samples can greatly help ML models become more effective in extracting biologically relevant patterns.
Such instances underline the fact that continuous collaboration with domain experts and the sharing of well-annotated data is needed to generate robust datasets in the future.
Ideally, structure in the composite datasets under study will be aligned with variables of interest, such as phenotype labels if available; if instead samples from the same cohort tend to group together regardless of phenotype, revisiting the construction of the dataset is warranted.
In the next section, we will discuss approaches that can aid in identifying and visualizing structure in datasets.
Box 1: Understanding experimental design of machine learning to inform requirements for data
Components of ML experiments
Machine learning algorithms identify patterns that explain or fit a given dataset.
Every machine learning algorithm goes through a training phase where it identifies underlying patterns in a given dataset to create a “trained” algorithm (a model) , and a testing phase where the model applies the identified patterns to unseen data points.
Typically, a machine learning algorithm is provided with the following fundamental parts as input: 1. a training dataset , 2. an evaluation dataset , 3. a held-out validation dataset .
These input data can be images, text, numbers, or other types of data which are typically encoded as a numerical representation of the input data.
A training dataset is used to expose the model to underlying patterns among the features present in the data of interest.
An evaluation dataset is a small test dataset which is used during the training phase to help the model iteratively update its parameters (i.e., hyperparameter_tuning or model tuning).
In many cases, a large training set may be subdivided to form a smaller training dataset and the evaluation dataset, both of which are used to train the model (see next section for more details on cross-validation).
In the testing phase, a new or unseen test dataset or held-out validation set is used to test whether the patterns learned by the model hold true in new data (are generalizable).
While the evaluation dataset helps the model iteratively update its parameters to learn important patterns in the training data, the held-out validation set helps test the generalizability of the model.
Generalizability of a model is its ability to recognize patterns that can help predict the class or an outcome for previously unseen data.
High generalizability of a model on previously unseen data suggest that the model has identified fundamental patterns in the data that may also inform our knowledge regarding the question of interest for which the experiment was designed.
Generalizability can be affected if data leakage occurs during training of the model, i.e., if a model is exposed to the same or similar data points in both the training set and the held-out test set.
Ensuring absence of any overlap or relatedness among data points or samples (e.g., samples with familial relationship, samples from same patient) used in the training set and held-out test set is important to avoid data leakage during model training.
Specifically in cases of rare genetic diseases where many samples can contain familial relationships, special care should be taken while crafting the training and testing sets to ensure that no data leakage occurs and the trained model has high generalizability.
Training and testing
The implementation of a machine learning experiment begins with splitting a single dataset of interest such that 90% of the data is used for training (generally subdivided into the training dataset and the evaluation dataset), and remaining 10% of the data is used for testing or validation (as the held-out validation dataset).
This makes sure that all the datasets involved in training and testing a model maintain uniformity in the features.
In case of rare diseases where multiple datasets may be combined to make a large enough training dataset, special care is taken to standardize the features and the patterns therein.
The iterative training stage helps the model learn important patterns in the training dataset and then use the evaluation dataset to test for errors in prediction and update its learning parameters (hyperparameter tuning).
The method by which the evaluation dataset tests the performance of the trained model and helps update the hyperparameters is called cross-validation.
To maximally utilize the available data for cross-validation, there can be multiple approaches to form the training and evaluation datasets e.g. leave-p-out cross-validation, leave-one-out cross-validation, k-fold cross-validation, Monte-Carlo random subsampling cross-validation [11].
In case of k-fold cross-validation, a given dataset is shuffled randomly and split into k parts.
One of the k parts is reserved as the evaluation dataset and the rest are cumulatively used as the training dataset.
In the next iteration, a different part is used as the evaluation dataset, while the rest are used for training.
Once the model has iterated through all k parts of the training and evaluation datasets, it is ready to be tested on the held-out validation dataset.
The held-out validation dataset is exposed to the model only once to estimate the accuracy of the model.
High accuracy of a model on the training dataset but low accuracy on the held-out dataset is a sign that the model has become overfit to the training set and has low generalizability.
If this is encountered, the experimenter is advised to revisit the dataset construction to make sure they meet the best practices outlined above.
Learning representations from rare disease data
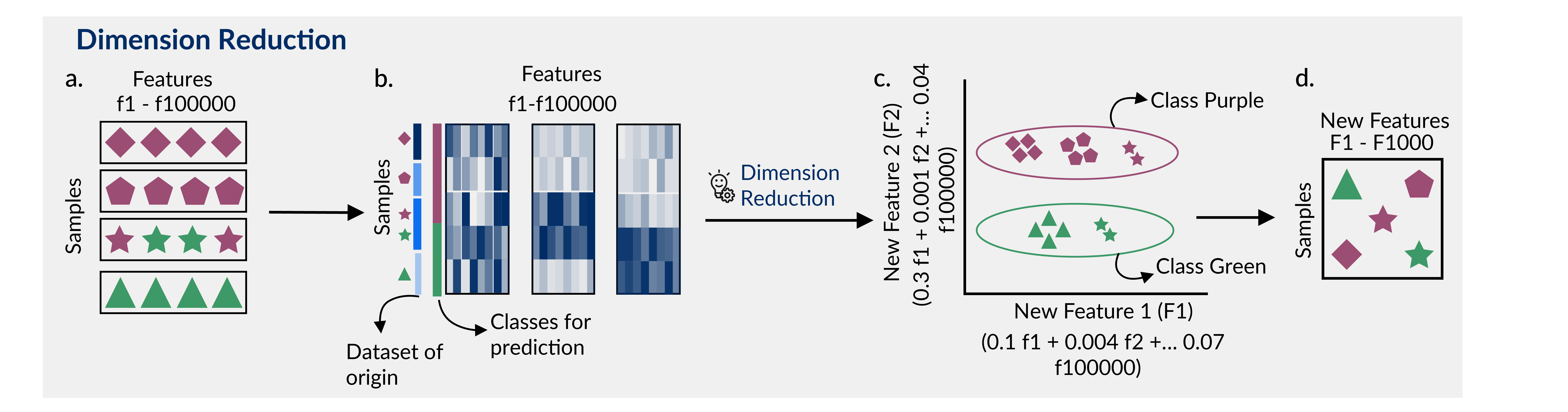
Dimensionality reduction methods like multidimensional scaling (MDS), principal components analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and uniform manifold approximation and projection (UMAP) can help ‘compress’ information from a large number of features into a smaller number of features in an unsupervised manner [12,13,14] (Figure 1C).
These methods not only help in reducing the number of features in various types of data [16,17], but can also be used to visualize structure or artifacts in the data (e.g. [18]), to define sample subgroups (e.g. [19], or for feature selection and extraction during application of specific machine learning models [20] (Figure 1D).
Methods like PCA, MDS, t-SNE, and UMAP can successfully identify the effect of these variables on the original data, though t-SNE and UMAP may require tuning of hyperparameters that may effect the output [14,21].
Furthermore, testing multiple dimensionality reduction methods, rather than a single method, may be necessary to obtain a more comprehensive portrait of the data [22].
Nguyen and Holmes discuss additional important considerations for using dimensionality reduction methods such as selection criteria and interpretation of results [23].
Beyond dimensionality reduction, other unsupervised learning approaches such as k-means clustering or hierarchical clustering have been used to characterize the structure present in genomic or imaging data [24,25].
Dimensionality reduction, or more fundamentally, representation learning, learns low-dimensional representations (composite features) from the raw data.
For example, representation learning through matrix factorization can extract features from transcriptomics datasets that are made of combinations of gene expression values found in the training data [26], and use them to interpret test data [22,27].
To ensure that the learned representations are generalizable to other data, the features learned by the model can be constrained through methods like regularization [28].
Representation learning generally requires many samples when applied to complex biological systems and therefore may appear to aggravate the curse of dimensionality.
However, it can be a powerful tool to learn low-dimensional patterns from large datasets and then find those patterns in smaller, related datasets.
In later sections, we will discuss this method of leveraging large datasets to reduce dimensionality in smaller datasets, also known as feature-representation-transfer learning.
Figure 1: Dimension reduction can help manage the curse of dimensionality in rare disease data. A) Multiple datasets (shapes) with multiple phenotypes (purple, green) are combined for an analysis. The data (e.g., transcriptomic data) are highly dimensional, having thousands of features (f1-f100000). B) Evaluating the features, it appears that a combination of features (e.g., expressed genes) partition the purple samples from the green samples. C) Applying a dimensionality reduction method (e.g., PCA) condenses these features into new features (e.g., New Feature 1, a combination of f1, f2 …. f100000, and New Feature 2, a different combination of f1, f2 …. f100000). New Feature 1 describes the difference in input dataset (shapes) while New Feature 2 describes the difference in phenotype (color). D) New features (F1-F1000) can be used to interrogate the biology of the input samples, develop classification models, or use other analytical techniques that would have been more difficult with the original dataset dimensions.
Manage model complexity while preserving the value of machine learning
Machine learning methods are generally accompanied by a few critical assumptions.
Assumption 1, the dataset contains equal number of samples for all classes in a dataset (i.e. no imbalance among classes).
2. the dataset is complete with adequate samples for all features (lack of sparsity).
3. there is no ambiguity about the labels for the samples in the dataset (i.e. no label-noise, applicable for supervised learning methods).
Rare disease datasets, however, violate many of these assumptions.
There is generally a high class imbalance due to small number of samples for specific classes (e.g., only a few patients with a particular rare disease in a health records dataset), abundant label-noise due to incomplete understanding of the disease, and in many cases sparsity in data.
All of these contribute to low signal to noise ratio in rare disease datasets.
Thus, applying ML to rare disease data without any accommodations for the above shortcomings may lead to models that lack stability required for reproducibility or simplicity required for interpretation.
In this section, we highlight a few ML techniques that can help manage the challenges in applying ML models applied to rare disease data.
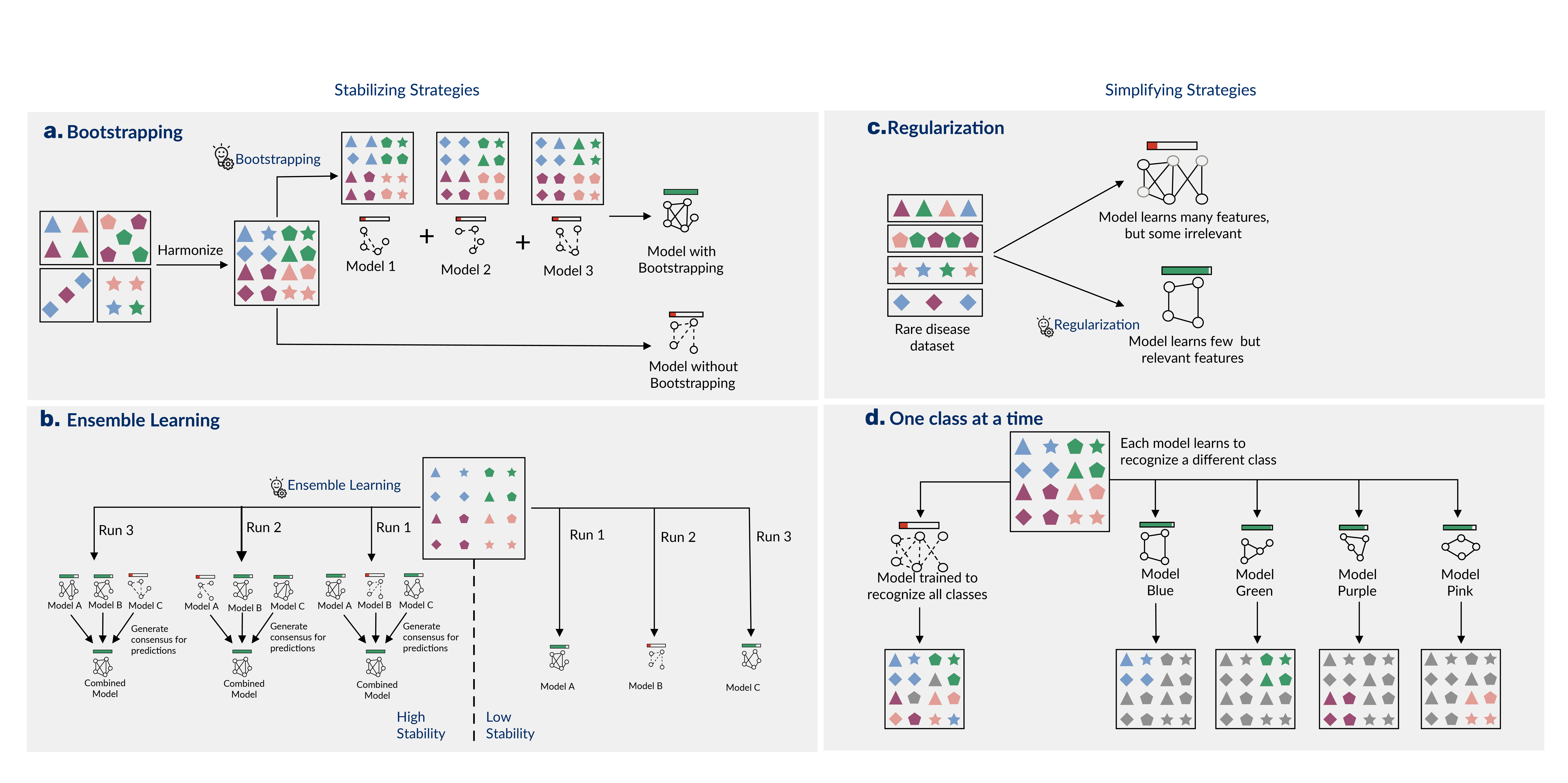
Accommodating class imbalance in datasets and increasing the stability of ML predictions can be achieved through decision tree-based ensemble learning methods (Figure[2]A-B).
Random forests use resampling (with replacement) based techniques like bagging (bootstrap aggregation) of independent decision trees to form a consensus about the important predictive features [30,31,32,33,34].
Additional approaches like combining random forests with resampling without replacement can generate confidence intervals for the model predictions by iteratively exposing the models to incomplete datasets, mimicking real world cases where most rare disease datasets are incomplete [35].
Recent studies suggest that there are limitations to decision tree-based ensemble methods when applied to rare disease datasets with substantial class imbalance and label-noise [36,37].
This has led to the adoption of cascade learning, a variant of ensemble learning, where multiple methods leveraging distinct underlying assumptions are used in tandem; and augmented with algorithms like AdaBoost (boosting) to capture stable patterns existing in silver standard data [38,39,40].
For example, a cascade learning approach for identifying rare disease patients from electronic health record data utilized independent steps for feature extraction (word2vec [41]), preliminary prediction with ensembled decision trees, and prediction refinement using data similarity metrics [37].
Combining these three methods resulted in better performance than other methods when implemented on the silver standard dataset in isolation.
Other approaches like inverse class weighting, oversampling (with methods like Synthetic Minority Oversampling Technique, abbreviated SMOTE), or cascade learning approaches that adequately represent the rarer classes are also beneficial in increasing stability of predictions while accommodating for class imbalance.
Presence of label-noise and sparsity in the data can lead to overfitting of models to the training data, such that the models show high prediction accuracy on training data but low prediction accuracy on encountering new data points.
Features identified as predictive by overfit models are often misrepresented in the training dataset making the model not generalizable to new data. [42]
Regularization can not only protect ML models from poor generalizability caused by overfitting, but also reduce model complexity by reducing the feature space available for training. (Figure[2]C)
Regularization is often used in rare variant discovery and immune cell signature discovery studies which are useful examples that also need to accommodate sparsity of data like rare diseases.
For example, among various regularization methods (ridge regression, LASSO regression, and elastic-net)[43], hybrid applications of LASSO for capturing combinations of rare variants have proven beneficial [44,45] .
In the context of rare immune cell signature discovery, elastic-net regression was found to outperform other regression approaches [43,46,47,48].
Thus regularization methods like LASSO or elastic-net have been methods of choice while working with rare observations; use of these regularization approaches should be considered while working with rare disease datasets.
Figure 2: Strategies to simplify models and stabilize predictions preserve the value of machine learning in rare disease. A-B) Strategies to build confidence in model predictions; A) A schematic showing the concept of bootstrap, B) A schematic showing the concept of ensemble learning to converge on reliable models; C-D) Strategies to simplify models by penalizing complexity in ML models; C) A schematic showing the concept of regularization to selectively learn relevant features, D) A schematic showing the concept of one-class-at-a-time learning to select few features at a time. Horizontal bars represent health of a model, models are represented as a network of nodes (features) and edges (relationships), nodes with solid edges represent real patterns, nodes with broken edges represent spurious patterns
Build upon prior knowledge and indirectly related data
Rare diseases often lack large, normalized datasets, limiting our ability to study key attributes of these diseases.
One strategy to overcome this is to integrate and explore rare disease information alongside other knowledge by combining a variety of different data types.
By using several data modalities (such as curated pathway, genetic data, or other data types), it may be possible to gain a better understanding of rare diseases (e.g., identifying novel genotype-phenotype relationships or opportunities for drug repurposing).
Knowledge graphs (KGs) which integrate related-but-different data types, create a rich multimodal data source (e.g., Monarch Graph Database [49], hetionet [50], PheKnowLator [51], and the Global Network of Biomedical Relationships [52], Orphanet [53]).
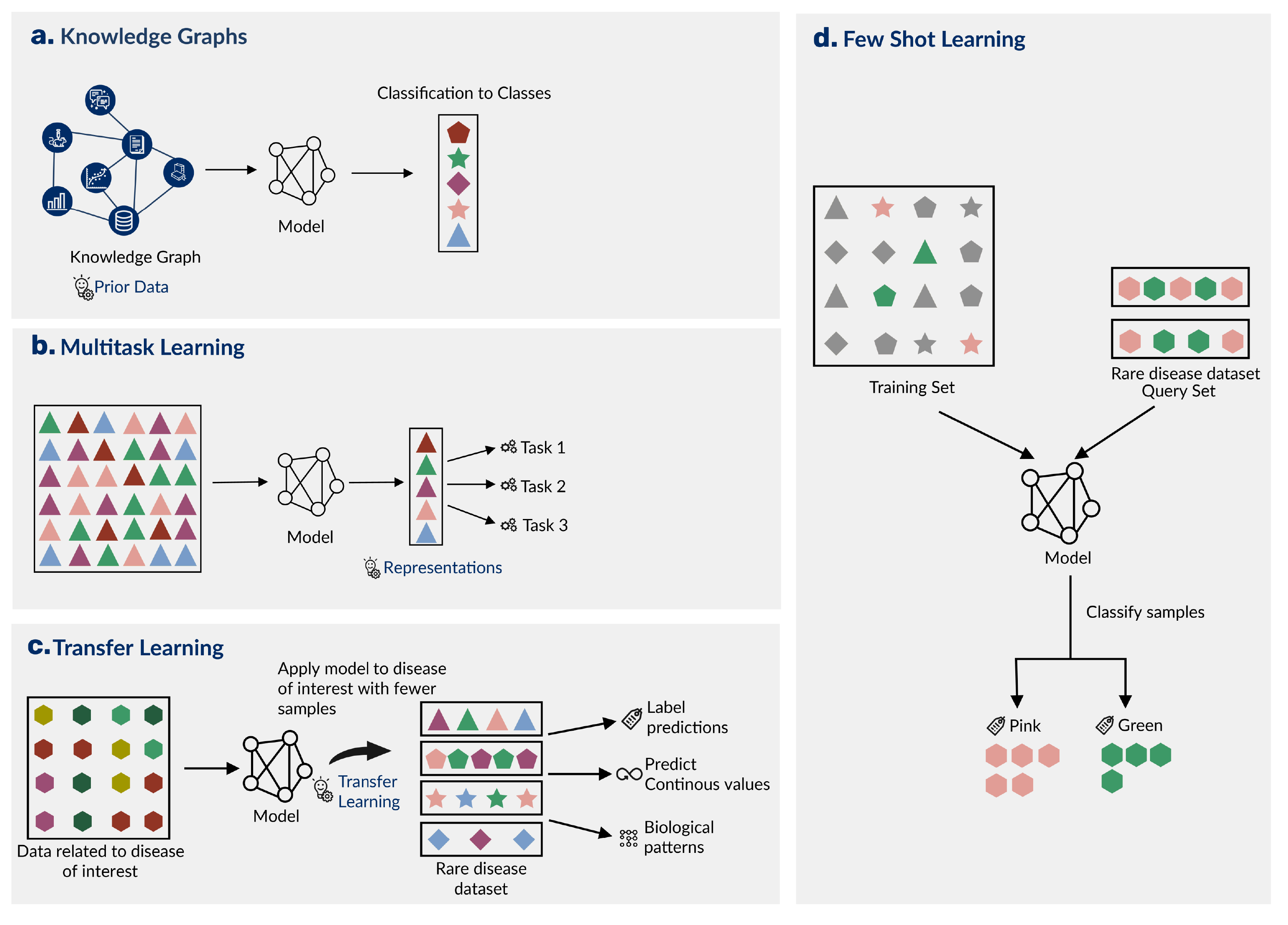
These graphs connect genetic, functional, chemical, clinical, and ontological data to enable the exploration of relationships of data with disease phenotypes through manual review [54] or computational methods [55,56]. (Figure[3]a)
KGs may include links or nodes that are specific to the rare disease of interest (e.g., an FDA approved treatment would be a specific disease-compound link in the KG) as well as links that are more generalized (e.g., gene-gene interactions noted in the literature for a different disease).
Rare disease researchers can repurpose general (i.e., not rare disease-specific) biological or chemical knowledge graphs to answer rare disease-based research questions [57].
There are a variety of tactics to sift through the large amounts of complex data in knowledge graphs.
One such tactic is to calculate the distances between nodes of interest (e.g., diseases and drugs to identify drugs for repurposing in rare disease [57]); this is often done by determining the “embeddings” (linear representations of the position and connections of a particular point in the graph) for nodes in the knowledge graph, and calculating the similarity between these embeddings.
Testing a variety of different methods to calculate node embeddings that can generate actionable insights for rare diseases is an active area of research [57], and an opportunity for continued research.
Another application of KGs is to augment or refine a dataset [58].
For example, Li et. al.[56] used a KG to identify linked terms in a medical corpus from a large number of patients, some with rare disease diagnoses.
They were able to augment their text dataset by identifying related terms in the clinical text to map them to the same term - e.g., mapping “cancer” and “malignancy” in different patients to the same clinical concept.
With this augmented and improved dataset, they were able to train and test a variety of text classification algorithms to identify rare disease patients within their corpus.
Finally, another possible tactic for rare disease researchers is to take a knowledge graph, or an integration of several knowledge graphs, and apply neural network-based algorithms optimized for graph data, such as a graph convolutional neural network.
Rao and colleagues [59] describe the construction of a KG using phenotype information (Human Phenotype Ontology) and rare disease information (Orphanet) and curated gene interaction/pathway data (Lit-BM-13, WikiPathways) [TODO: citations for other resources - HPO, etc].
They then trained a spectral graph convolution neural network on this KG to identify and rank potentially causal genes for the Orphanet rare diseases, and were able to use this model to predict causal genes for a ground truth dataset of rare diseases with known causal genes.
While several groups have already published on the use of KGs to study rare diseases, we expect that the growth of multi-modal datasets and methods to analyze KGs will make them a more popular and important tool in the application of ML in rare disease.
Another approach that builds on prior knowledge and large volumes of related data is transfer learning.
Transfer learning leverages shared features, e.g., normal developmental processes that are aberrant in disease or an imaging anomaly present in both rare and common diseases, to advance our understanding of rare diseases.
Transfer learning, where a model trained for one task or domain (source domain) is applied to another related task or domain (target domain), can be supervised or unsupervised.
Among various types of transfer learning, feature-representation-transfer approaches learn representations from the source domain and apply them to a target domain [60] (Figure[3]b).
That is, representation learning, as discussed in an earlier section, does not need to be applied only to describe the dataset on which the algorithm was trained – it can also be used to elucidate signals in sufficiently similar data.
For instance, low-dimensional representations can be learned from tumor transcriptomic data and transferred to describe patterns associated with genetic alterations in cell line data [22].
In the next section, we will summarize specific instances of applying transfer learning, along with other techniques described herein, to the study of rare diseases.
Figure 3: Strategies that build upon prior knowledge help ML models learn patterns in rare disease datasets. A) Knowledge graphs integrate different data types and may allow models to learn from connections that are rare disease-specific or happen in many biomedical contexts. B) Transfer learning is when a model trained in for one task or domain is applied to another, related task. C) Multitask learning uses models that learn and leverage shared representations to predict multiple, related tasks. D) Few-shot learning generalizes a previously trained model to predict a new, related task with a limited number of samples.
Using composite approaches can be a powerful strategy
We have described multiple approaches for maximizing the success of ML applications in rare disease, but it is rarely sufficient to use any of these techniques in isolation.
Below, we highlight two recent works in the rare disease domain that draw on concepts of feature-representation-transfer, use of prior data, and regularization.
A large public dataset of acute myeloid leukemia (AML) patient samples with no drug response data and a small in vitro experiment with drug response data form the basis of our first example [61].
Training an ML model on the small in vitro dataset alone faced the curse of dimensionality and the dataset size prohibited representation learning.
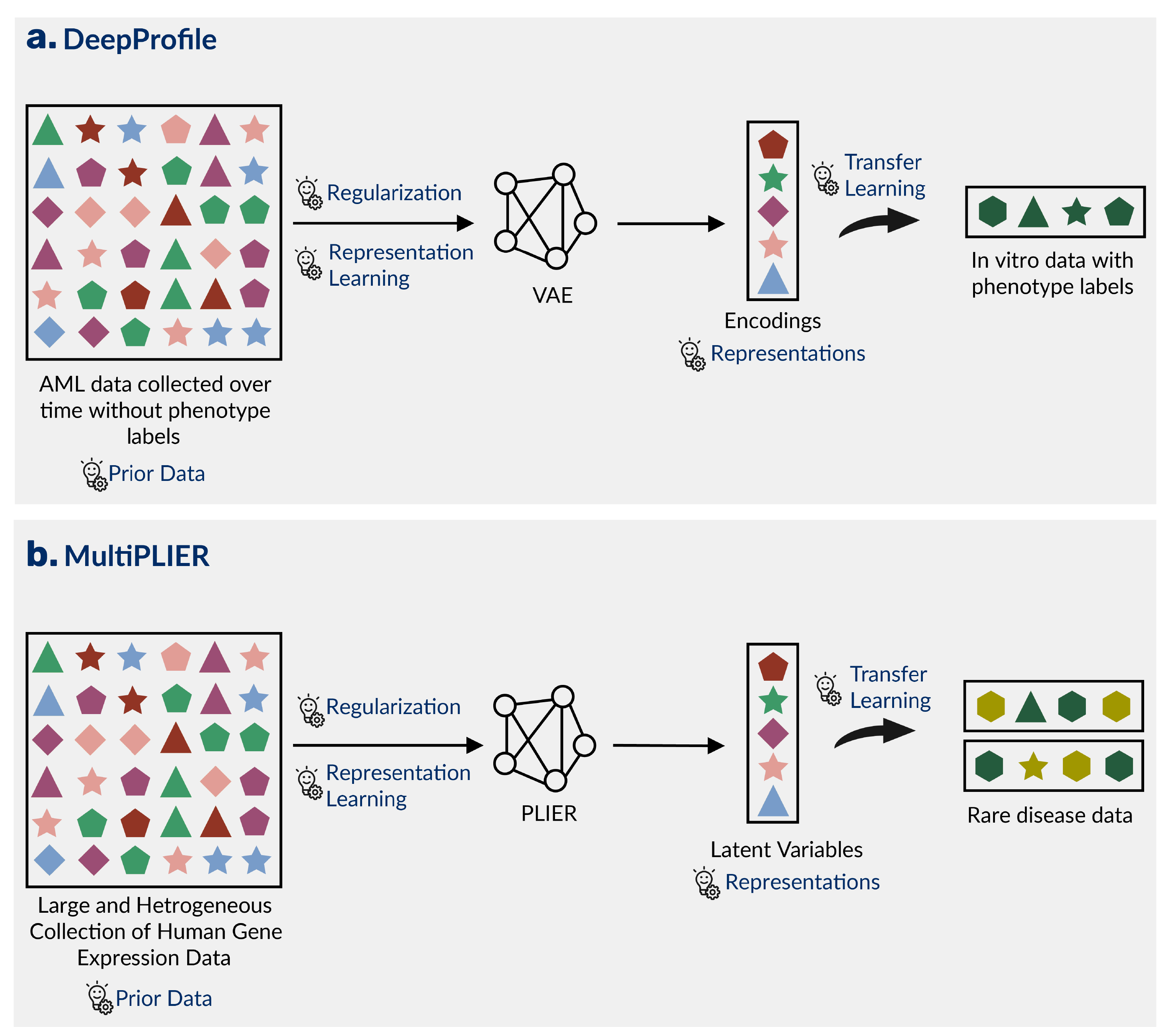
Dincer et al. trained a variational autoencoder on the large AML patient dataset (VAE; see definitions) to learn meaningful representations in an approach termed DeepProfile [62] (Figure[4]a).
The representations or encodings learned by the VAE were then transferred to the small in vitro dataset reducing it’s number of features from thousands to eight, and improving the performance of the final LASSO linear regression model.
In addition to improvement in performance, the encodings learned by the VAE captured more biological pathways than PCA, which may be attributable to the constraints on the encodings imposed during the training process (see definitions).
Similar results were observed for prediction of histopathology in another rare cancer dataset [62].
While DeepProfile was centered on training on an individual disease and tissue combination, some rare diseases affect multiple tissues that a researcher may be interested in studying together for the purpose of biological discovery.
Studying multiple tissues poses significant challenges and a cross-tissue analysis may require comparing representations from multiple models.
Models trained on a low number of samples may learn representations that “lump together” multiple biological signals, reducing the interpretability of the results.
To address these challenges, Taroni et al. trained a Pathway-Level Information ExtractoR (PLIER) (a matrix factorization approach that takes prior knowledge in the form of gene sets or pathways) on a large generic collection of human transcriptomic data [63].
PLIER used constraints (regularization) that learned latent variables aligned with a small number of input gene sets, making it suitable for biological discovery or description of rare disease data.
The authors transferred the representations or latent variables learned by the model to describe transcriptomic data from the unseen rare diseases antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and medulloblastoma in an approach termed MultiPLIER [64]. (Figure[4]b)
MultiPLIER used one model to describe multiple datasets instead of reconciling output from multiple models, thus making it possible to identify commonalities among disease manifestations or affected tissues.
DeepProfile [62] and MultiPLIER [64] exemplify modeling approaches that can incorporate prior knowledge – thereby constraining the model space according to plausible or expected biology – or that can share information across datasets.
These two methods capitalize on the fact that similar biological processes are observed across different biological contexts and that the methods underlying the approaches can effectively learn about those processes.
Figure 4: Combining multiple strategies strengthens the performance of ML models in rare disease. A) The authors of DeepProfile trained a variational autoencoder (VAE) to learn a representation from acute myeloid leukemia data without phenotype labels, transferred those representations to a small dataset with phenotype labels, and found that it improved prediction performance [62]. B) The authors of MultiPLIER trained a Pathway-Level Information ExtractoR (PLIER) model on a large, heterogeneous collection of expression data and transferred the representations to multiple datasets from unseen rare diseases [63].
Outlook
Throughout this perspective, we highlighted various challenges in applying ML methods to rare disease data as well as examples of approaches that address these challenges.
Small sample size, while significant, is not the only roadblock towards application of ML in rare disease data.
The high dimensionality of modern data requires creative approaches, such as learning new representations of the data, to manage the curse of dimensionality.
Leveraging prior knowledge and transfer learning methods to appropriately interpret data is also required.
Furthermore, we posit that researchers applying machine learning methods on rare disease data should use techniques that increase confidence (i.e., bootstrapping) and penalize complexity of the resultant models (i.e., regularization) to enhance the generalizability of their work.
All of the approaches highlighted in this perspective come with weaknesses that may undermine investigators’ confidence in using these techniques for rare disease research.
We believe that the challenges in applying ML to rare disease are opportunities for data generation and method development going forward.
In particular, we identify the following two areas as important for the field to explore to increase the utility of machine learning in rare disease.
Emphasis on not just “more n” but “more meaningful n”
Mindful addition of data is key for powering the next generation of analysis in rare disease data.
While there are many techniques to collate rare data from different sources, low-quality data may hurt the end goal even if it adds to the size of the dataset.
In our experience, collaboration with domain experts has proved to be critical in gaining insight into potential sources of variation in the datasets.
An anecdotal example from the authors’ personal experience: conversations with a rare disease clinician revealed that samples in a particular tumor dataset were collected using vastly different surgical techniques (laser ablation and excision vs standard excision).
This information that was not readily available to non-experts, but was obvious the clinician.
Such instances underline the fact that continuous collaboration with domain experts and the sharing of well-annotated data is needed to generate robust datasets in the future.
In addition to sample scarcity, there is a dearth of comprehensive phenotypic-genotypic databases in rare disease.
While rare disease studies that collect genomic and phenotypic data are becoming more common [65,66,67], an important next step is to develop comprehensive genomics-based genotype-phenotype databases that prioritize clinical and genomics data standards in order to fuel interpretation of features extracted using ML methods.
Finally, mindful sharing of data with proper metadata and attribution to enable prompt data reuse is of utmost important in building datasets that can be of great value in rare disease [68].
Development of methods that reliably support mechanistic interrogation of specific rare diseases
The majority of ML methods for rare disease that we have investigated are applied to classification tasks.
Conversely, we’ve found few examples of methodologies that interrogate biological mechanisms of rare diseases.
This is likely a consequence of a dearth of methods that can tolerate the constraints imposed by rare disease research such as phenotypic heterogeneity and limited data.
An intentional push towards developing methods or analytical workflows that address this will be critical to apply machine learning approaches to rare disease data.
Method development with rare disease applications in mind requires the developers to bear the responsibility of ensuring that the resulting model is trustworthy.
The field of natural language processing has a few examples of how this can be achieved [69].
One way to increase trust in a developed model is by helping users understand the behavior of the developed model through providing explanations regarding why a certain model made certain predictions [69].
Another approach is to provide robust error analysis for newly developed models to help users understand the strengths and weaknesses of a model [70,71,72].
Adoption of these approaches into biomedical ML is quickly becoming necessary as machine learning approaches become mainstream in research and clinical settings.
Finally, methods that can reliably integrate disparate datasets will likely always remain a need in rare disease research.
To facilitate such analyses in rare disease, methods that rely on finding structural correspondences between datasets (“anchors”) may be able to transform the status-quo of using machine learning methods in rare disease [73,74,75].
We speculate that this an important and burgeoning area of research, and we are optimistic about the future of applying machine learning approaches to rare diseases.
Definitions
VAE:
Variational Autoencoders or VAEs are unsupervised neural networks that use hidden layers to learn or encode representations from available data while mapping the input data to the output data.
VAEs are distinct from other autoencoders since the distribution of the encodings are regularized such that they are close to a normal distribution, which may contribute to learning more biologically relevant signals [22].
References
1. The use of machine learning in rare diseases: a scoping review
Julia Schaefer, Moritz Lehne, Josef Schepers, Fabian Prasser, Sylvia Thun Orphanet Journal of Rare Diseases (2020-06-09) https://doi.org/ghb3wx
DOI: 10.1186/s13023-020-01424-6 · PMID: 32517778 · PMCID: PMC7285453
2. Learning statistical models of phenotypes using noisy labeled training data
Vibhu Agarwal, Tanya Podchiyska, Juan M Banda, Veena Goel, Tiffany I Leung, Evan P Minty, Timothy E Sweeney, Elsie Gyang, Nigam H Shah Journal of the American Medical Informatics Association (2016-11) https://doi.org/f9bxf9
DOI: 10.1093/jamia/ocw028 · PMID: 27174893 · PMCID: PMC5070523
3. Classification in the Presence of Label Noise: A Survey
Benoit Frenay, Michel Verleysen IEEE Transactions on Neural Networks and Learning Systems (2014-05) https://doi.org/f5zdgq
DOI: 10.1109/tnnls.2013.2292894 · PMID: 24808033
6. The properties of high-dimensional data spaces: implications for exploring gene and protein expression data
Robert Clarke, Habtom W. Ressom, Antai Wang, Jianhua Xuan, Minetta C. Liu, Edmund A. Gehan, Yue Wang Nature Reviews Cancer (2008-01) https://doi.org/ffksnf
DOI: 10.1038/nrc2294 · PMID: 18097463 · PMCID: PMC2238676
13. Principal component analysis: a review and recent developments
Ian T. Jolliffe, Jorge Cadima Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences (2016-04-13) https://doi.org/gcsfk7
DOI: 10.1098/rsta.2015.0202 · PMID: 26953178 · PMCID: PMC4792409
14. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction
Leland McInnes, John Healy, James Melville arXiv:1802.03426 [cs, stat] (2020-09-17) http://arxiv.org/abs/1802.03426
19. Dimensionality reduction by UMAP to visualize physical and genetic interactions
Michael W. Dorrity, Lauren M. Saunders, Christine Queitsch, Stanley Fields, Cole Trapnell Nature Communications (2020-03-24) https://doi.org/ggqcqp
DOI: 10.1038/s41467-020-15351-4 · PMID: 32210240 · PMCID: PMC7093466
22. Compressing gene expression data using multiple latent space dimensionalities learns complementary biological representations
Gregory P. Way, Michael Zietz, Vincent Rubinetti, Daniel S. Himmelstein, Casey S. Greene Genome Biology (2020-05-11) https://doi.org/gg2mjh
DOI: 10.1186/s13059-020-02021-3 · PMID: 32393369 · PMCID: PMC7212571
24. Clustering cancer gene expression data: a comparative study
Marcilio CP de Souto, Ivan G Costa, Daniel SA de Araujo, Teresa B Ludermir, Alexander Schliep BMC Bioinformatics (2008-11-27) https://doi.org/dqqbn6
DOI: 10.1186/1471-2105-9-497 · PMID: 19038021 · PMCID: PMC2632677
25. Removing Batch Effects From Histopathological Images for Enhanced Cancer Diagnosis
Sonal Kothari, John H. Phan, Todd H. Stokes, Adeboye O. Osunkoya, Andrew N. Young, May D. Wang IEEE Journal of Biomedical and Health Informatics (2014-05) https://doi.org/gdm9jd
DOI: 10.1109/jbhi.2013.2276766 · PMID: 24808220 · PMCID: PMC5003052
26. Deriving disease modules from the compressed transcriptional space embedded in a deep autoencoder
Sanjiv K. Dwivedi, Andreas Tjärnberg, Jesper Tegnér, Mika Gustafsson Nature Communications (2020-02-12) https://doi.org/gg7krm
DOI: 10.1038/s41467-020-14666-6 · PMID: 32051402 · PMCID: PMC7016183
27. CoGAPS: an R/C++ package to identify patterns and biological process activity in transcriptomic data
Elana J. Fertig, Jie Ding, Alexander V. Favorov, Giovanni Parmigiani, Michael F. Ochs Bioinformatics (2010-11-01) https://doi.org/cwqsv4
DOI: 10.1093/bioinformatics/btq503 · PMID: 20810601 · PMCID: PMC3025742
28. Regularized Machine Learning in the Genetic Prediction of Complex Traits
Sebastian Okser, Tapio Pahikkala, Antti Airola, Tapio Salakoski, Samuli Ripatti, Tero Aittokallio PLoS Genetics (2014-11-13) https://doi.org/ghrqhq
DOI: 10.1371/journal.pgen.1004754 · PMID: 25393026 · PMCID: PMC4230844
29. Review and evaluation of penalised regression methods for risk prediction in low‐dimensional data with few events
Menelaos Pavlou, Gareth Ambler, Shaun Seaman, Maria De Iorio, Rumana Z Omar Statistics in Medicine (2015-10-29) https://doi.org/ggn9zg
DOI: 10.1002/sim.6782 · PMID: 26514699 · PMCID: PMC4982098
31. Evaluating predictive modeling algorithms to assess patient eligibility for clinical trials from routine data
Felix Köpcke, Dorota Lubgan, Rainer Fietkau, Axel Scholler, Carla Nau, Michael Stürzl, Roland Croner, Hans-Ulrich Prokosch, Dennis Toddenroth BMC Medical Informatics and Decision Making (2013-12-09) https://doi.org/f5jqvh
DOI: 10.1186/1472-6947-13-134 · PMID: 24321610 · PMCID: PMC4029400
33. Utilising artificial intelligence to determine patients at risk of a rare disease: idiopathic pulmonary arterial hypertension
David G. Kiely, Orla Doyle, Edmund Drage, Harvey Jenner, Valentina Salvatelli, Flora A. Daniels, John Rigg, Claude Schmitt, Yevgeniy Samyshkin, Allan Lawrie, Rito Bergemann Pulmonary Circulation (2019-11) https://doi.org/gg4jc7
DOI: 10.1177/2045894019890549 · PMID: 31798836 · PMCID: PMC6868581
35. Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways that Define Tumor Heterogeneity in NF1
Jineta Banerjee, Robert J Allaway, Jaclyn N Taroni, Aaron Baker, Xiaochun Zhang, Chang In Moon, Christine A Pratilas, Jaishri O Blakeley, Justin Guinney, Angela Hirbe, … Sara JC Gosline Genes (2020-02-21) https://doi.org/gg4rbj
DOI: 10.3390/genes11020226 · PMID: 32098059 · PMCID: PMC7073563
36. Enhancing techniques for learning decision trees from imbalanced data
Ikram Chaabane, Radhouane Guermazi, Mohamed Hammami Advances in Data Analysis and Classification (2019-03-02) https://doi.org/ghz4sz
DOI: 10.1007/s11634-019-00354-x
37. Learning to Identify Rare Disease Patients from Electronic Health Records.
Rich Colbaugh, Kristin Glass, Christopher Rudolf, Mike Tremblay Volv Global Lausanne Switzerland AMIA … Annual Symposium proceedings. AMIA Symposium (2018-12-05) https://www.ncbi.nlm.nih.gov/pubmed/30815073
PMID: 30815073 · PMCID: PMC6371307
38. Component-based face detection
B. Heiselet, T. Serre, M. Pontil, T. Poggio Institute of Electrical and Electronics Engineers (IEEE) (2005-08-25) https://doi.org/c89p2b
DOI: 10.1109/cvpr.2001.990537
39. The Architecture of the Face and Eyes Detection System Based on Cascade Classifiers
Andrzej Kasinski, Adam Schmidt Advances in Soft Computing (2007) https://doi.org/cbzq9n
DOI: 10.1007/978-3-540-75175-5_16
40. Real time facial expression recognition with AdaBoost
Yubo Wang, Haizhou Ai, Bo Wu, Chang Huang Institute of Electrical and Electronics Engineers (IEEE) (2004) https://doi.org/crv3sq
DOI: 10.1109/icpr.2004.1334680
41. Efficient Estimation of Word Representations in Vector Space
Tomas Mikolov, Kai Chen, Greg Corrado, Jeffrey Dean arXiv (2013-09-10) https://arxiv.org/abs/1301.3781
42. Definitions, methods, and applications in interpretable machine learning
W. James Murdoch, Chandan Singh, Karl Kumbier, Reza Abbasi-Asl, Bin Yu Proceedings of the National Academy of Sciences (2019-10-29) https://doi.org/ggbhmq
DOI: 10.1073/pnas.1900654116 · PMID: 31619572 · PMCID: PMC6825274
43. Regularization and variable selection via the elastic net
Hui Zou, Trevor Hastie Journal of the Royal Statistical Society: Series B (Statistical Methodology) (2005-04) https://doi.org/b8cwwr
DOI: 10.1111/j.1467-9868.2005.00503.x
44. Methods for Detecting Associations with Rare Variants for Common Diseases: Application to Analysis of Sequence Data
Bingshan Li, Suzanne M. Leal The American Journal of Human Genetics (2008-09) https://doi.org/d4jpcb
DOI: 10.1016/j.ajhg.2008.06.024 · PMID: 18691683 · PMCID: PMC2842185
45. Comparison of statistical approaches to rare variant analysis for quantitative traits
Han Chen, Audrey E Hendricks, Yansong Cheng, Adrienne L Cupples, Josée Dupuis, Ching-Ti Liu BMC Proceedings (2011-11-29) https://doi.org/b9mf4x
DOI: 10.1186/1753-6561-5-s9-s113 · PMID: 22373209 · PMCID: PMC3287837
46. Regularized logistic regression with adjusted adaptive elastic net for gene selection in high dimensional cancer classification
Zakariya Yahya Algamal, Muhammad Hisyam Lee Computers in Biology and Medicine (2015-12) https://doi.org/f73xvj
DOI: 10.1016/j.compbiomed.2015.10.008 · PMID: 26520484
47. Sparse logistic regression with a L1/2 penalty for gene selection in cancer classification
Yong Liang, Cheng Liu, Xin-Ze Luan, Kwong-Sak Leung, Tak-Ming Chan, Zong-Ben Xu, Hai Zhang BMC Bioinformatics (2013-06-19) https://doi.org/gb8v2x
DOI: 10.1186/1471-2105-14-198 · PMID: 23777239 · PMCID: PMC3718705
48. An elastic-net logistic regression approach to generate classifiers and gene signatures for types of immune cells and T helper cell subsets
Arezo Torang, Paraag Gupta, David J. Klinke BMC Bioinformatics (2019-08-22) https://doi.org/gg5hmj
DOI: 10.1186/s12859-019-2994-z · PMID: 31438843 · PMCID: PMC6704630
49. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species
Christopher J. Mungall, Julie A. McMurry, Sebastian Köhler, James P. Balhoff, Charles Borromeo, Matthew Brush, Seth Carbon, Tom Conlin, Nathan Dunn, Mark Engelstad, … Melissa A. Haendel Nucleic Acids Research (2017-01-04) https://doi.org/f9v7bz
DOI: 10.1093/nar/gkw1128 · PMID: 27899636 · PMCID: PMC5210586
50. Systematic integration of biomedical knowledge prioritizes drugs for repurposing
Daniel Scott Himmelstein, Antoine Lizee, Christine Hessler, Leo Brueggeman, Sabrina L Chen, Dexter Hadley, Ari Green, Pouya Khankhanian, Sergio E Baranzini eLife (2017-09-22) https://doi.org/cdfk
DOI: 10.7554/elife.26726 · PMID: 28936969 · PMCID: PMC5640425
51. A Framework for Automated Construction of Heterogeneous Large-Scale Biomedical Knowledge Graphs
Tiffany J. Callahan, Ignacio J. Tripodi, Lawrence E. Hunter, William A. Baumgartner Cold Spring Harbor Laboratory (2020-05-02) https://doi.org/gg338z
DOI: 10.1101/2020.04.30.071407
54. Structured reviews for data and knowledge-driven research
Núria Queralt-Rosinach, Gregory S Stupp, Tong Shu Li, Michael Mayers, Maureen E Hoatlin, Matthew Might, Benjamin M Good, Andrew I Su Database (2020) https://doi.org/ggsdkj
DOI: 10.1093/database/baaa015 · PMID: 32283553 · PMCID: PMC7153956
55. Learning Drug-Disease-Target Embedding (DDTE) from knowledge graphs to inform drug repurposing hypotheses
Changsung Moon, Chunming Jin, Xialan Dong, Saad Abrar, Weifan Zheng, Rada Y. Chirkova, Alexander Tropsha Journal of Biomedical Informatics (2021-07) https://doi.org/gmpgs6
DOI: 10.1016/j.jbi.2021.103838 · PMID: 34119691
56. Improving rare disease classification using imperfect knowledge graph
Xuedong Li, Yue Wang, Dongwu Wang, Walter Yuan, Dezhong Peng, Qiaozhu Mei BMC Medical Informatics and Decision Making (2019-12-05) https://doi.org/gg5j65
DOI: 10.1186/s12911-019-0938-1 · PMID: 31801534 · PMCID: PMC6894101
57. A Literature-Based Knowledge Graph Embedding Method for Identifying Drug Repurposing Opportunities in Rare Diseases
Daniel N. Sosa, Alexander Derry, Margaret Guo, Eric Wei, Connor Brinton, Russ B. Altman World Scientific Pub Co Pte Ltd (2019-12) https://doi.org/gmpgs7
DOI: 10.1142/9789811215636_0041
58. Rare disease knowledge enrichment through a data-driven approach
Feichen Shen, Yiqing Zhao, Liwei Wang, Majid Rastegar Mojarad, Yanshan Wang, Sijia Liu, Hongfang Liu BMC Medical Informatics and Decision Making (2019-02-14) https://doi.org/gm48cq
DOI: 10.1186/s12911-019-0752-9 · PMID: 30764825 · PMCID: PMC6376651
59. Phenotype-driven gene prioritization for rare diseases using graph convolution on heterogeneous networks
Aditya Rao, Saipradeep VG, Thomas Joseph, Sujatha Kotte, Naveen Sivadasan, Rajgopal Srinivasan BMC Medical Genomics (2018-07-06) https://doi.org/gnb3q7
DOI: 10.1186/s12920-018-0372-8 · PMID: 29980210 · PMCID: PMC6035401
60. A Survey on Transfer Learning
Sinno Jialin Pan, Qiang Yang IEEE Transactions on Knowledge and Data Engineering (2010-10) https://doi.org/bc4vws
DOI: 10.1109/tkde.2009.191
61. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia
Su-In Lee, Safiye Celik, Benjamin A. Logsdon, Scott M. Lundberg, Timothy J. Martins, Vivian G. Oehler, Elihu H. Estey, Chris P. Miller, Sylvia Chien, Jin Dai, … Pamela S. Becker Nature Communications (2018-01-03) https://doi.org/gcpx72
DOI: 10.1038/s41467-017-02465-5 · PMID: 29298978 · PMCID: PMC5752671
62. DeepProfile: Deep learning of cancer molecular profiles for precision medicine
Ayse Berceste Dincer, Safiye Celik, Naozumi Hiranuma, Su-In Lee Cold Spring Harbor Laboratory (2018-05-26) https://doi.org/gdj2j4
DOI: 10.1101/278739
63. Pathway-level information extractor (PLIER) for gene expression data
Weiguang Mao, Elena Zaslavsky, Boris M. Hartmann, Stuart C. Sealfon, Maria Chikina Nature Methods (2019-06-27) https://doi.org/gf75g6
DOI: 10.1038/s41592-019-0456-1 · PMID: 31249421 · PMCID: PMC7262669
64. MultiPLIER: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease
Jaclyn N. Taroni, Peter C. Grayson, Qiwen Hu, Sean Eddy, Matthias Kretzler, Peter A. Merkel, Casey S. Greene Cell Systems (2019-05) https://doi.org/gf75g5
DOI: 10.1016/j.cels.2019.04.003 · PMID: 31121115 · PMCID: PMC6538307
65. Rare-disease genetics in the era of next-generation sequencing: discovery to translation
Kym M. Boycott, Megan R. Vanstone, Dennis E. Bulman, Alex E. MacKenzie Nature Reviews Genetics (2013-09-03) https://doi.org/ghvhsd
DOI: 10.1038/nrg3555 · PMID: 23999272
66. Paediatric genomics: diagnosing rare disease in children
Caroline F. Wright, David R. FitzPatrick, Helen V. Firth Nature Reviews Genetics (2018-02-05) https://doi.org/gcxbr8
DOI: 10.1038/nrg.2017.116 · PMID: 29398702
67. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders
David R. Adams, Christine M. Eng New England Journal of Medicine (2018-10-04) https://doi.org/gf49m7
DOI: 10.1056/nejmra1711801 · PMID: 30281996
69. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier
Marco Ribeiro, Sameer Singh, Carlos Guestrin Association for Computational Linguistics (ACL) (2016) https://doi.org/gg8ggh
DOI: 10.18653/v1/n16-3020
70. Errudite: Scalable, Reproducible, and Testable Error Analysis
Tongshuang Wu, Marco Tulio Ribeiro, Jeffrey Heer, Daniel Weld Association for Computational Linguistics (ACL) (2019) https://doi.org/ggb9kk
DOI: 10.18653/v1/p19-1073
73. Domain Adaptation with Structural Correspondence Learning
John Blitzer, Ryan McDonald, Fernando Pereira Proceedings of the 2006 Conference on Empirical Methods in Natural Language Processing (2006-07) https://aclanthology.org/W06-1615
74. Heterogeneous domain adaptation using manifold alignment
Chang Wang, Sridhar Mahadevan Proceedings of the Twenty-Second international joint conference on Artificial Intelligence - Volume Volume Two (2011-07-16) https://dl.acm.org/doi/10.5555/2283516.2283652
ISBN: 9781577355144
75. Comprehensive Integration of Single-Cell Data
Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M. Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, Rahul Satija Cell (2019-06) https://doi.org/gf3sxv
DOI: 10.1016/j.cell.2019.05.031 · PMID: 31178118 · PMCID: PMC6687398
 0000-0002-1775-3645
·
0000-0002-1775-3645
·  jaybee84
jaybee84 allawayr
allawayr